Category: Structure
Distributing points uniformly on a sphere
Earlier this semester I had a project where I had to calculate the solvent-accessible surface area of a protein given its 3-dimensional structure (in a PDB file). The suggested method of implementation was to simulate the surface of each atom by distributing 500 points (roughly) evenly across the surface defined by its van der Waals radius. Although that sounds straightforward, it actually took me a while to figure out how to randomly sample points from a sphere defined by a point and a radius.
I did some extensive searching on Google, but most of the methods I found came with the caveat that there would be a greater concentration of points near the poles than at the equator. I finally came across this page, which offered a clear and concise method for sampling points from a sphere.
- Set up a 2-dimensional coordinate system: the z dimension (running from -R to R, where R is the van der Waals radius) and
(longitude, running from 0 to
radians)
- Generate random coordinates:
and
- Use the following relation (
) to calculate latitude (
):
- Finally, convert to Cartesian (xyz) coordinates
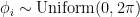
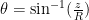

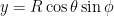

Repeat this process 500 times for each atom to simulate its surface!
Rodrigues’ rotation formula
I’m taking a structural informatics course this semester, and this is my first in-depth exposure to working with biomolecular structures. The class’ first assignment was to download a specific PDB file, parse the atom coordinates, and determine all sorts of bond lengths, bond angles, and torsion angles. This required me to review concepts I haven’t used much since linear algebra and multivariable calculus, but all in all this part wasn’t too hard.
The last part of the assignment was a kicker though. I had to select the 30th residue in the protein, set two of its dihredral angles to 0º (effectively rotating the remaining portion of the structure along bonds between backbone atoms), and recompute the new atomic coordinates. I had no idea where to start!
One of the other student’s in the class mentioned Rodrigues’ rotation formula, and after looking into things, it seemed to be the answer to our question. In a general sense, if you want to rotate given a vector 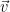


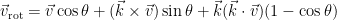
In the context of the homework assignment, 
F1 score and gene annotation comparisons
Burset and Guigó published a foundational paper on evaluating gene annotations in 1996. A lot of my work as a graduate student has involved writing software for comparing multiple sets of gene structure annotations against each other, and I’ve used the statistics described in this paper (matching coefficient, correlation coefficient, sensitivity, specificity) as the basis for my comparisons. However, there is another statistic (called the F1 score) that is (apparently) used commonly for analyzing gene annotations, and someone recently recommended that I should include this statistic in my comparisons. Never having heard of it, I decided to investigate.
I found several papers that referenced the F1 score (none of them were related to gene structure annotation, by the way) and was able to eventually track down the origin of the statistic. It was introduced by van Rijsbergen in 1979 in a text on information retrieval and has since found application in a variety of fields. The F1 score combines two other commonly used statistics: the precision 
With precision defined as

and recall defined as
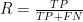
we now define the F1 score as follows.
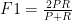
After 15 minutes of searching, 10 minutes of reading, and 5 minutes of coding, my software now has a new feature!
